- Medicines
- Personal Care
- Skin Cream
- Sunscreen
- Face Wash
- Skin and Body Soap
- Acne Care
- Body Lotions
- Moisturising Lotion
- Moisturising Cream
- Mosquito Repellent
- Moisturising Gel
- Body Wash
- Hair Oils
- Hair Shampoo
- Hair Conditioners
- Hair Supplements
- Hair Colour
- Hair Serum
- Hair Mask
- Hair Solutions
- Baby Diapers and Wipes
- Baby Lotion and Moisturising Cream
- Baby Bath Essentials
- Baby Skin Care
- Baby and Infant Food
- Baby Healthcare
- Women Multivitamins
- Ovulation Test Kit and Women Intimate Care
- Sanitary Pads
- Nutritional Drinks
- Condoms
- Lubricants
- Massage Gels
- Personal Body Massagers
- Men Performance Booster
- Sexual Health Supplements
- Massage Oils
- Ayurveda
- Tooth Paste
- Mouth Ulcer Gel
- Mouthwash
- Toothache and Gum Pain
- Tooth Brush
- Gargle Solution
- Orthopaedic Supports
- Adult Diapers
- Footwear
- Mobility and Support Accessories
- Urinary Support and Care
- Health Conditions
- Bone and Joint Care
- Digestive Care
- Eye Care
- Pain Relief
- Smoking Cessation
- Liver Care
- Stomach Care
- Cold and Cough
- Heart Care
- Kidney Care
- Piles, Fissures & Fistula
- Respiratory Care
- Mental Wellness
- Derma Care
- Pre and Probiotics
- Acidity
- Gas
- Constipation
- Loose Motion/Diarrhoea
- Digestive Fibres
- Digestive Enzymes
- Eye Lubricant Drops
- Lens Solution
- Safety Eye Wear
- Eye Cream
- Eye Vitamins and Supplements
- Eye Drops
- Eye Ointment and Gel
- Nicotine Patch
- Nicotine Gum
- Nicotine Lozenges
- Cough Syrups
- Chest Rubs and Balms
- Nasal Spray
- Lozenges
- Inhalant Capsules
- Cold and Cough Tablets
- Vitamins & Supplements
- Diabetes Care
- Healthcare Devices
- Homeopathic Medicine
- Health Guide
Cystic fibrosis
Cystic fibrosis is a genetic disorder that causes the body to produce thick, sticky mucus, leading to severe respiratory and digestive problems. It is an inherited condition caused by mutations in the cystic fibrosis transmembrane conductance regulator gene, typically diagnosed in childhood but sometimes not until adulthood. Treatment focuses on managing symptoms and preventing complications.
Last updated on : 27 Jun, 2025
Read time : 14 mins
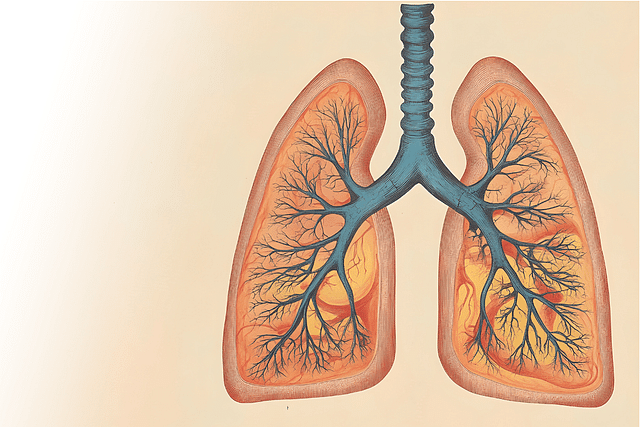
Overview of Disease
Cystic fibrosis is a life-threatening genetic disorder that primarily affects the lungs and digestive system. It is caused by a defective gene that leads to the production of thick, sticky mucus that clogs the airways and various organs. As a result, individuals with cystic fibrosis face a range of challenges, including difficulty breathing, recurring lung infections, and malnutrition. Despite advances in treatment, cystic fibrosis remains a serious condition that requires ongoing management and care.
What is Cystic fibrosis?
Cystic fibrosis, also known as mucoviscidosis, is an inherited disorder caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. This gene is responsible for regulating the movement of salt and water in and out of cells. When the CFTR gene is defective, it leads to the production of abnormally thick and sticky mucus that accumulates in the lungs, pancreas, and other organs. This mucus buildup disrupts normal function and leads to a range of symptoms and complications. Although cystic fibrosis is a progressive condition, advances in treatment have greatly improved the outlook for those affected.
Key Factors about Cystic fibrosis
| Category | Details |
| Also Referred as | Mucoviscidosis, Fibrocystic disease of pancreas, Cystic fibrosis of pancreas |
| Commonly Occurs In | People of Northern European ancestry. However, it can affect all racial and ethnic groups |
| Affected Organ | Lungs, Pancreas, Liver, Kidneys, Intestine, Sinuses, and Reproductive system |
| Type | Genetic disorder inherited in an autosomal recessive manner |
| Common Signs | Difficulty breathing, Coughing up mucus, Poor growth, Fatty stool, and Sinus infections |
| Consulting Specialist | Medical geneticist and pulmonologist |
| Treatement Procedures | Physiotherapy, Antibiotics, Pancreatic enzyme replacement, and CFTR modulators |
| Managed By | Antibiotics, anti-inflammatory medications, and CFTR modulators |
| Mimiciking Condition | Other respiratory and digestive disorders but distinct owing to genetic basis and specific diagnostic tests |
Symptoms of Cystic fibrosis
The symptoms of cystic fibrosis can vary in severity and may change over time. Some common symptoms include:
1. Respiratory symptoms
Persistent cough that produces thick mucus
Wheezing and shortness of breath
Recurring lung infections and inflammation
Chronic sinusitis and nasal congestion
2. Digestive symptoms
Diarrhoea and malnutrition
Poor growth and weight loss
Blockage of the intestine (meconium ileus) in newborns
Pancreatitis and cystic fibrosis-related diabetes mellitus (CFRDM) in adults
3. Other symptoms
Saltiness of sweat (parents may notice a salty taste when kissing their child)
Infertility in men owing to congenital bilateral absence of the vas deferens (CBAVD)
Complications in pregnancy for women with cystic fibrosis
Stages of Cystic fibrosis
The stages of cystic fibrosis can be broadly categorised as follows:
Newborns may present with meconium ileus, respiratory distress, and failure to thrive.
Children often experience recurring lung infections, digestive problems, and poor growth.
Adults with cystic fibrosis may face additional challenges, such as the development of diabetes, osteoporosis, and liver disease.
Causes of Cystic fibrosis
Cystic fibrosis primarily affects the lungs and digestive system. The primary cause of cystic fibrosis is a mutation in the CFTR gene, which is responsible for regulating the movement of salt and water in and out of cells.
Inheritance pattern: Cystic fibrosis is an autosomal recessive disorder, meaning an individual must inherit one copy of the mutated gene from each parent to develop the condition.
CFTR gene mutation: The CFTR gene mutation leads to the production of thick, sticky mucus that accumulates in various organs, particularly the lungs and pancreas.
Other factors that may influence the development and severity of cystic fibrosis include:
Ethnicity: Cystic fibrosis is more common among individuals of Northern European descent, although it can affect people of any race or ethnicity.
Environmental factors: Exposure to certain environmental triggers, such as cigarette smoke or air pollution, may exacerbate cystic fibrosis symptoms.
Risk Factors
Several factors can increase the risk of developing cystic fibrosis.
1. Genetic Factors
Cystic fibrosis is caused by a mutation in the CFTR gene. To develop cystic fibrosis, an individual must inherit two abnormal copies of this gene, one from each parent.
Carriers who have one abnormal copy of the CFTR gene do not have cystic fibrosis symptoms themselves, but they can pass the faulty gene on to their children. If both parents are carriers, each pregnancy has a 25% chance of the child having CF.
2. Family History
Having a family history of cystic fibrosis significantly raises the risk, especially if one or both biological parents either have cystic fibrosis themselves or are carriers of the abnormal gene.
3. Ethnicity
Cystic fibrosis is most prevalent in people of Northern European descent, particularly Caucasians. The condition is less common in Hispanic, African American, and Asian American populations.
4. Age of Diagnosis
While cystic fibrosis is usually diagnosed in early childhood, with over 75% of cases identified by age 2, some milder cases may not present until adulthood.
Complications
Cystic fibrosis can cause a range of complications, particularly as the condition progresses. Some of the main areas affected include:
1. Respiratory System
Recurring lung infections are common in cystic fibrosis because of the thick, sticky mucus that traps bacteria. Over time, repeated infections can damage the lungs.
Cystic fibrosis can cause bronchiectasis, a chronic condition where the airways become abnormally widened and scarred, further increasing infection risk.
Pneumothorax, or collapsed lung, may occur in advanced cystic fibrosis. This happens when damaged lung tissue ruptures, allowing air to leak into the chest cavity.
2. Digestive System
Mucus can block ducts in the pancreas, preventing digestive enzymes from reaching the intestines. This leads to malnutrition and poor growth.
Patients with cystic fibrosis have an increased risk of diabetes, gallstones, and liver disease owing to mucus buildup affecting organ function.
3. Reproductive System
Most men with cystic fibrosis are infertile owing to blocked or absent vas deferens, the tubes that carry sperm.
While women with CF may have thicker cervical mucus that makes conception more difficult, they can still carry successful pregnancies with proper care.
Prevention of Cystic fibrosis
As cystic fibrosis arises from genetic causes, prevention efforts are primarily centred around genetic counselling and testing.
1. Genetic Testing
Genetic tests can screen for mutations in the CFTR gene to identify cystic fibrosis carriers. Testing is especially useful for those with a family history of cystic fibrosis or couples planning a pregnancy.
These carrier tests analyse DNA from a blood, saliva, or cheek cell sample to detect abnormalities in the cystic fibrosis gene.
2. Pre-Conception Genetic Counselling
Couples who are cystic fibrosis carriers can seek genetic counselling before conceiving. This allows them to understand their risks and make informed choices about starting a family.
3. Prenatal Testing Options
For at-risk pregnancies, prenatal tests such as amniocentesis or chorionic villus sampling can determine if the foetus has inherited cystic fibrosis.
4. Lowering Environmental Risks
While not directly preventing mucoviscidosis, avoiding smoking and secondhand smoke exposure can help manage cystic fibrosis symptoms if present.
Maintaining overall health through proper nutrition and exercise is also beneficial.
Diagnosis & Tests
Early diagnosis is crucial for the effective management and treatment of cystic fibrosis. There are several diagnostic tests available to confirm the presence of the condition.
Newborn screening: In many countries, newborns are routinely screened for cystic fibrosis using a blood test that measures the level of immunoreactive trypsinogen (IRT).
Sweat test: The sweat test is considered the gold standard for diagnosing cystic fibrosis. It measures the amount of salt in a person's sweat, as individuals with cystic fibrosis have higher levels of salt in their sweat.
Genetic testing: Genetic tests can identify specific mutations in the CFTR gene, confirming the diagnosis of cystic fibrosis and helping to determine the severity of the condition.
In addition to these tests, a comprehensive evaluation by a multidisciplinary team at a specialised cystic fibrosis care centre is essential for an accurate diagnosis and the development of an individualised treatment plan. This evaluation may include a thorough medical history, physical examination, and additional tests to assess the function of various organs affected by cystic fibrosis.
Treatment & Management
Cystic fibrosis (CF) requires a comprehensive, lifelong treatment strategy focused on maintaining lung function, managing digestive issues, and preventing complications.
- Respiratory Therapy:
Airway clearance techniques like chest physiotherapy, postural drainage, and the use of devices such as oscillatory vests help remove thick mucus from the lungs. Inhaled medications are also key components:
- Salbutamol: A short-acting bronchodilator that relaxes the airway muscles, improving airflow and easing breathing in cystic fibrosis patients.
- Hypertonic saline: Inhaled solution that helps draw water into the airways, thinning mucus and making it easier to clear from the lungs.
- Dornase alfa: A mucolytic enzyme that breaks down DNA in thick mucus, reducing its viscosity and improving lung function in cystic fibrosis.
- Tobramycin: An inhaled antibiotic that targets Pseudomonas aeruginosa infections commonly seen in cystic fibrosis, helping to control chronic lung infections.
- Aztreonam: An inhaled antibiotic used to treat lung infections caused by gram-negative bacteria, especially Pseudomonas aeruginosa, in cystic fibrosis patients.
- Anti-inflammatory Medications:
- Ibuprofen helps reduce lung inflammation in children.
- Azithromycin is used for its anti-inflammatory and antibacterial properties.
- Nutritional Support:
A high-calorie, high-fat diet supports energy needs. Pancreatic enzyme replacement therapy (pancrelipase) is essential for digestion and nutrient absorption. Fat-soluble vitamin supplements (A, D, E, and K) are also routinely prescribed.
- Other Medications:
- Insulin if CF-related diabetes develops
- Proton pump inhibitors (e.g., omeprazole) may be used to enhance enzyme function and manage reflux
Management of CF is guided by a multidisciplinary team, and treatment plans are tailored to the individual's symptoms, genotype, and age. Regular monitoring and early intervention are crucial to improving outcomes and extending life expectancy.
Living with Disease
Living with cystic fibrosis requires a comprehensive and ongoing management plan to mitigate the symptoms and complications of this genetic disorder. While there is no cure for cystic fibrosis, a combination of daily management techniques, medications, and lifestyle adjustments can significantly improve quality of life.
1. Daily Management
Proper nutrition is crucial for individuals with cystic fibrosis. A high-calorie, high-fat diet helps maintain weight and support growth. Pancreatic enzyme supplements and vitamins are often necessary to aid digestion and ensure adequate nutrient absorption. Regular physical activity is also essential for overall health and to help clear mucus from the lungs. Exercise and pulmonary rehabilitation can be particularly beneficial in managing cystic fibrosis symptoms.
2. Medications and Therapies
Airway clearance therapy is a cornerstone of cystic fibrosis treatment. Techniques such as chest physical therapy, inhalers, and nebulisers help loosen and clear the thick, sticky mucus that characterises mucoviscidosis. Antibiotics and anti-inflammatory medicines are frequently used to treat and prevent lung infections, which are common in cystic fibrosis owing to the buildup of mucus in the airways.
3. Lifestyle Adjustments
To minimise the risk of infections, people with cystic fibrosis should avoid close contact with others who are sick and practice diligent hand hygiene. Stress management techniques, such as relaxation exercises, can help with improving overall well-being and coping with the emotional challenges of living with a chronic condition.
4. Social and Emotional Support
Joining support groups, either online or in-person, can provide invaluable connections and resources for individuals with cystic fibrosis and their families. Organizations specifically working with cystic fibrosis specialists can also assist in navigating insurance, legal, and financial issues related to living with cystic fibrosis.
When to See a Doctor?
Managing cystic fibrosis involves regular monitoring and prompt medical intervention when necessary. Knowing when to call the doctor is essential for preventing complications and maintaining optimal health.
1. Signs of Infection
Fever is often an indicator of infection in individuals with cystic fibrosis. If you develop a fever, it is crucial to notify your doctor immediately. Similarly, any changes in cough or sputum production, particularly if the mucus becomes thicker or more discoloured, should be reported promptly.
2. Respiratory Complications
Shortness of breath or difficulty breathing are red flags that require immediate medical attention. These symptoms may indicate a worsening of cystic fibrosis-related lung disease or the development of a serious complication. Chest pain or signs of a collapsed lung (pneumothorax) are medical emergencies that necessitate urgent care.
3. Gastrointestinal Issues
Severe abdominal pain or vomiting may signal a gastrointestinal blockage or other intestinal complication of cystic fibrosis. If you experience these symptoms, seek medical advice without delay.
Key Takeaways
Cystic fibrosis, or mucoviscidosis, is a complex genetic disorder that affects multiple organ systems. While the symptoms and severity of cystic fibrosis vary from person to person, understanding the key aspects of this condition is essential for effective management.
Cystic fibrosis is caused by mutations in the CFTR gene, which lead to the production of thick, sticky mucus that accumulates in the lungs, digestive system, and other organs. Common symptoms include persistent coughing, frequent lung infections, difficulty breathing, poor growth and weight gain, and digestive problems.
Although there is no cure for cystic fibrosis, a multi-faceted treatment approach can significantly improve outcomes and quality of life. This includes daily airway clearance techniques, medications to manage infections and reduce inflammation, nutritional support, and lifestyle modifications to prevent complications.
Early diagnosis through newborn screening and prompt initiation of treatment are critical for optimising health outcomes in individuals with cystic fibrosis. Regular follow-up with a multidisciplinary team experienced in cystic fibrosis care is essential for monitoring disease progression, adjusting treatments, and addressing any complications that may arise.
Significant progress has been made in understanding the genetic basis of cystic fibrosis and developing targeted therapies. Continued research efforts aim to identify new treatments, improve existing therapies, and ultimately find a cure for this life-limiting disorder.
FAQs
What is the primary reason behind the development of cystic fibrosis?
Cystic fibrosis is caused by a genetic mutation in the CFTR gene, which is inherited from both parents and affects salt and water movement in cells.
How long can individuals with cystic fibrosis expect to live?
Owing to advancements in medical treatments, people with cystic fibrosis now have an average life expectancy extending into their mid to late 30s, with some living even longer.
In what ways does cystic fibrosis impact an individual's daily life?
Cystic fibrosis significantly affects a person's life by causing breathing difficulties, frequent lung infections, digestive issues, malnutrition, and various complications, necessitating regular medical care and symptom management.
Can you list three typical signs and symptoms of cystic fibrosis?
Three characteristic symptoms of cystic fibrosis include persistent cough and lung infections due to thick mucus, digestive problems leading to malnutrition, and chronic sinusitis with recurrent infections.
Is there a permanent cure available for cystic fibrosis?
While there is currently no cure for cystic fibrosis, various treatments are available to manage symptoms, prevent complications, and slow disease progression, including medications, therapies, and ongoing research into gene therapy.
At what age do people typically develop cystic fibrosis?
Cystic fibrosis is a genetic disorder present from birth, with symptoms usually appearing in early childhood, although some individuals may be diagnosed later in life.
Does cystic fibrosis affect men and women equally?
Cystic fibrosis affects both men and women equally, as it is an autosomal recessive genetic disorder not linked to gender.
References
- National Heart, Lung, and Blood Institute. (n.d.). Cystic fibrosis. https://www.nhlbi.nih.gov/health-topics/cystic-fibrosis
- Davies, J. C., Alton, E. W., & Bush, A. (2007). Cystic fibrosis. BMJ (Clinical research ed.), 335(7632), 1255–1259. https://doi.org/10.1136/bmj.39391.713229.AD
- Chen, Q., Shen, Y., & Zheng, J. (2021). A review of cystic fibrosis: Basic and clinical aspects. Animal Models and Experimental Medicine, 4(3), 220–232. https://doi.org/10.1002/ame2.12180
- Dickinson, K. M., & Collaco, J. M. (2021). Cystic fibrosis. Pediatrics in Review, 42(2), 55–67. https://doi.org/10.1542/pir.2019-0212
Check Related Salts
Browse Other Conditions
Latest health articles







Top Health Essentials





























Disclaimer
Top-Selling Medicines:
...View more
Top-OTC medicines:
...View more
Company
About UsHealth ArticleHealth StoriesHealth LibraryDiseases & Health ConditionsAyurvedaUnderstanding Generic MedicinesAll MedicinesAll BrandsNeed HelpFAQSecuritySavings CalculatorSubscribe
Registered Office Address
Grievance Officer
Download Truemeds
Contact Us
Our customer representative team is available 7 days a week from 9 am - 9 pm.
v4.20.1
2026 - Truemeds | All rights reserved. Our content is for informational purposes only. See additional information.
Our Payment Partners

